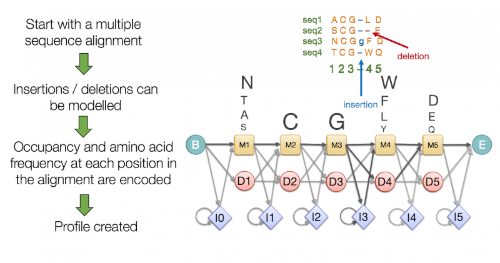
The Hidden Markov Model (HMM), a statistical method introduced to the literature in the 70s, was first used for speech recognition, then and today, it is trendy in the analysis of biological sequences such as protein sequences and DNA sequences [1]. The main idea of the model is that it is a finite model that describes a probability distribution over an infinite number of possible sequences [2]. This model can detect and classify a sequence using a given motif profile in its use in bioinformatics. It creates scoring by aligning with the profile and the desired motif, and thanks to this scoring, it reaches the correct classification result [3]. In bHLHDB, classification is made in accordance with PFAM protocols with the HMM package named HMMER [4] prepared with Python Programming Language. This package runs from the bHLH.hmm profile file from PFAM and classifies the bHLH TF from the protein sequences supplied to it.
References
[1]: M. Franzese and A. Iuliano, "Hidden Markov Models," Encyclopedia of Bioinformatics and Computational Biology, vol. 1, p. 753-762, 2019.
[2]: S. R. Eddy, "Hidden Markov Models," Current Opinion in Structural Biology, vol. 6, p. 361-365, 1996.
[3]: A. Krogh, "An Introduction to Hidden Markov Models for Biological Sequences," in In Computational Methods in Molecular Biology, Elsevier, 1998, pp. 46-63.
[4]: http://hmmer.org
image: https://en.wikipedia.org/wiki/HMMER